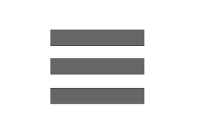

 structure of the charge agglomeration at very high dielectric constant. (b) percolated structure of charges at low high electric constant. As dielectric constant decreases, electrostatic strengths become stronger thereby causing stronger agglomeration. (c) Experimental proof showing counterion hopping as a mechanism of charge transport. (d) Trajectories of counterions showing hopping transport between two electrodes.")
Organic electrolyte batteries are widely used for practical energy storage, however due to fast discharge and thermal problems among others, polymer electrolyte-based batteries are currently being considered as possible next generation power sources for portable electronics. In this project we have focused on the fundamental physics of charge and counterion transport in model polymer electrolyte systems. The material that was used for this purpose is also known as an ionomer electrolyte because of the very low charge density on the backbone of the polymer chain. As a first step, we have already performed coarse-grained Molecular Dynamics (CG-MD) simulations to develop understanding of the effects of the dielectric constant on the microphase separation and counterion transport of this class of ionomer electrolytes The CG-MD results allow us to connect the "dots" between structural inhomogeneity and counterion hopping in ionomer electrolytes at a molecular level thereby providing guidelines for the design of better materials for future technologies. Using the program SASENA, we also calculated the dynamic structure factors (I(q,t)) for counterions showing a slowdown of counterion relaxation with decreasing dielectric constant. We also began working on a fully atomistic MD study (this enables capturing dynamic processes with shorter time and length scales) for ionomer electrolytes using LAMMPS (Large Scale Atomic/Molecular Massively Parallel Simulator) which will be compared with neutron experiments. We have constructed polyethylene (PE) oxide polymers in a highly concentrated solution and then replaced the -oxygen- with acrylic acid (AA) monomer. This allows a fully atomistic model of PE-AA to be generated which can be used as an input to LAMMPS.
We used different force fields (FF) between different bonded and non-bonded atomic interactions and equilibrated the structures to a melt state. Those studies show morphological details, however, the dynamics require more careful observation. There are certain numerical/physics issues in the fully atomistic model that needs to be addressed before a conclusive result can be obtained. A test simulation of a polyethylene melt using the Dreiding force field equilibrates well and the statistical quantities were calculated that match well with previous results. It should be noted the Dreiding force field is united atom not a fully atomistic FF, therefore additional work needed towards a fully atomistic simulation is currently under way.

