

Andrey Kovalevsky
Contact: kovalevskyay@ornl.gov or (505) 310-4184Research
Living organisms depend on the action of enzymes that catalyze chemical reactions too slow to support life. Enzymes are central in fighting disease, with most current clinical drugs targeting human, bacterial, or viral enzymes. In biotechnology, utilization of enzymes is the “green” option for biofuel and chemical production. Understanding the remarkable catalytic efficiency of enzymes in mechanistic terms has been the goal of biochemistry for decades, and the knowledge has had profound effects on biotechnological processes and drug design. Science is still working toward this elusive goal, demanding understanding at the level of atoms and electrons. Our approach to study enzymes combines innovative experimental and theoretical methodologies, including X-ray and neutron crystallography, neutron spectroscopy, and molecular simulations, in a novel way, providing mechanistic views of enzymes with unprecedented detail and then applying this knowledge to protein engineering and drug design.
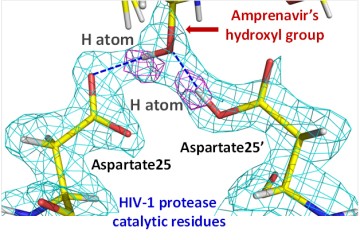
- Mechanistic, drug binding and drug resistance studies of the human immunodeficiency virus type 1 (HIV-1) protease and its precursors.
- Mechanistic studies of reactivation of organophosphate-conjugated human acetylcholinesterase (hAChE) by oximes and the design of novel accelerated reactivators.
- Mechanistic studies of vitamin B6 (PLP)-dependent enzymes.