
The phenomenon of crystallization is still not completely understood. Even though instrumentation and user facilities for Structural Biology have seen dramatic advancements in recent years, making the data collection and processing trouble-free and leaving the actual crystallization of a biomacromolecule to represent the major pitfall en route to a three-dimensional structure. While in case of X-ray protein crystallography one usually needs to "just" crystallize a protein, for neutron protein crystallography the need is transferred into a different realm: one has to grow large crystals, which are generally orders of magnitude bigger than those required for X-ray diffraction. This task sometimes turns out to be insurmountable, because making the leap to much larger crystals can be extremely exigent. Frequently, obtaining a protein crystal that is satisfactory to conduct a neutron diffraction experiment relies more on an act of serendipity than on the skills and background knowledge of a researcher. In this chapter we will outline the crystallization methodologies that have been developed (in some cases quite recently) to produce large protein crystals, and the techniques of H/D exchange in the crystals with heavy water (D2O).
Protein sample preparation. Considerations for sample preparation are similar for both X-ray and neutron crystallography, although they may differ in some details. Protein has to be over 95% pure, because the presence of contaminants can considerably hamper crystal growth. It is beneficial to centrifuge the protein solution at speeds exceeding 30,000g employing, for example, an air-driven ultracentrifuge in order to rid of insoluble particulates including oil droplets; however, more normal speeds of 16,000-23,000g would suffice for many protein preparations. Protein concentration (expressed as [protein] heretofore) is another crucial factor for the success in growing larger crystals, because it is the amount of solute present in a crystallization drop that generally limits the size of crystals, provided other factors like precipitant concentration and the number of nucleation sites are not a concern. Consequently, initial [protein] before the sample is mixed with precipitant should be as high as the protein solubility permits. (N.B. For very soluble proteins there is no gain for subsequent crystallization efforts if [protein] exceeds 100 mg/mL). To reach the highest possible [protein] in a sample one can concentrate the protein by centrifuging it in a concentrator containing a specific molecular weight cut-off membrane until the protein starts to precipitate. The [protein] remaining in the solution after spinning down the precipitate would represent the highest attainable concentration. This way one can be absolutely sure that the amount of the protein in the crystallization drop is sufficient. In some cases, achieving this goal would require careful investigation of the protein solubility profile and, perhaps, use of low concentrations of NaCl or KCl (0.2-0.4M) to keep one's protein in solution and avoid the salting-in effect. (N.B. The use of glycerol should be avoided, where possible, because it acts as the inhibitor of crystal growth)
Crystal growth techniques. One important conclusion from the above considerations is that a crystallization drop set up for producing large crystals has to be large as well. For the majority of protein crystallizations for neutron protein crystallography the drops should be in the range of 200-1000 mL. As a result, obviously, the hanging-drop method, which is employed almost universally in manual setup for growing X-ray quality crystals, is impractical for large crystal growth, since the largest hanging drop that can be produced is about 50 mL. Batch and sitting-drop, and to a lesser extent dialysis, methods have been routinely utilized for obtaining crystals
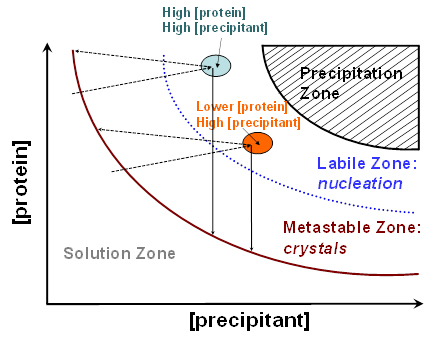
Figure 1. Idealized phase diagram of a protein solution as a function a precipitant concentration. Areas with different [protein] and [precipitant], that are depicted as color-filled ovals. Solutions with high [protein] and high [precipitant] are believed to give the largest crystals, which is always true for vapor diffusion methods (light cyan oval). For the batch method, it was found that solutions with lower [protein] and slightly higher [precipitant] can produce similarly large crystals without resorting to using large amounts of the precious protein sample (orange oval). The vertical solid arrows illustrate the shift in [protein] during crystal growth with batch method. Inclined dashed arrows depict the shift in [protein] during crystal growth with sitting drop method.
of the neutron diffraction quality. Figure 1 depicts the idealized phase diagram of a protein solution and contains the precipitation, labile, metastable and solution zones. The labile and metastable zones together represent a supersaturated solution. Nucleation that precedes crystal growth occurs in the labile zone, effectively shifting the [protein] into the metastable zone where the crystal growth ensues; the latter process further removes the protein from the liquid phase and the crystals stop growing when [protein] reaches the solubility curve. The largest crystals can always be obtained if the [protein] and [precipitant] are quite high, placing these conditions into the light cyan oval on the graph. For batch method, these concentrations are achieved immediately at the moment of mixing the protein and precipitant solutions. For sitting-drop method, they are reached during the water vapor diffusion process. For many proteins, however, working with such high concentrations means that a lot of the precious sample is consumed in just a few crystallization drops. It turns out that similar quality crystals can be grown by reducing the starting [protein] by 1.5-to-2 fold from the highest-achievable concentration (N.B. The final [protein] in a crystallization drop would depend on the exact conditions used). With this methodology, up to a half of the protein is saved making it possible to set up almost twice as many crystallization drops. This technique was applied for obtaining neutron diffraction quality crystals of the deoxy-human normal adult hemoglobin (Chatake et al., 2007), xylose isomerase (Kovalevsky et al., 2010a), equine cyanomet hemoglobin (Kovalevsky et al., 2010b) and carbonic anhydrase (Fisher et al., 2010). Best crystals can be obtained when the two-dimensional solubility diagram is constructed based on the crystallization conditions that have been formulated to give good crystals, with [protein] in the y-axis and the [precipitant] in the x-axis, as has been described for urate oxidase (Budayova-Spano et al., 2006). Furthermore, if a robotic liquid handling system is utilized the whole phase diagram can be scanned relatively easily by setting up 96-well microbatch (under oil) or sitting drop crystallizations. Generally, the software supplied with many robotic systems has the capability to calculate the crystallization conditions, with fine or coarse concentration steps, by supplying the computer program with the ranges of final (i.e., in the drop) [protein] and [precipitant]. (N.B. When crystallization drops are scaled up the [precipitant] may have to be slightly tweaked to higher or lower values).
Various reservoirs, commercial or custom-made, can be utilized for holding the crystallization drop itself and the mother liquor, when necessary. Nonetheless, perhaps the best setup for both the batch and the vapor diffusion techniques is available from Hampton Research Corp. (Aliso Viejo, CA, USA) in the form of the sandwich box kit, consisting of the siliconized 9-well plates, plastic supports and the sandwich boxes. The 9-well plates are ideal for crystallizing proteins using the batch method. Each well can hold up to 1mL of liquid, although drops should not exceed ~600mL for batch crystallizations. Use of parafin oil should be avoided, because the oil will inadvertently end up coating the crystal and will add to the incoherent background of scattered neutrons originating from the numerous hydrogen atoms of the oil's hydrocarbon molecules. Instead, the 9-well plate surface can be sealed air-tight with the CrystalClear tape, also supplied by Hampton Research Corp. (N.B. The wells of the 9-well plate should be treated with a siliconizing reagent, e.g. Sigmacot available from Sigma-Aldrich Co. (St. Louis, MO, USA), after each cleaning and before performing new crystallization trials in order to always maintain the pristine surface of the glass well). The vapor diffusion crystallizations can also be straightforwardly made with the sandwich box setup. The sandwich box is filled with 50-100 mL of the mother liquor and the 9-well plate is placed on top of the plastic support inside the box. (N.B. A layer of vacuum grease should be applied to the sandwich box walls and the cover to ensure the air-tight closure of the setup).
Crystallization by dialysis is a variation of the vapor diffusion technique, in which the [protein] and [precipitant] inside the dialysis button increase due to slow diffusion of the water out of the sample into the mother liquor and counter-diffusion of the precipitant in reverse manner through a semi-permeable membrane. The membrane should have a specific molecular weight cut-off so that the protein is kept inside the dialysis button during crystal growth. Dialysis is the only approach that can be applied to growing crystals by lowering, rather than increasing, the [precipitant]. Sometimes crystals can be obtained by dialyzing the sample against pure H2O or D2O due to the salting-in effect. Nowadays, dialysis buttons can be bought in different sizes allowing one to set up drops of up to 350 mL in volume. The method of dialysis has been successfully applied for growing neutron diffraction quality crystals of D-xylose isomerase (Hanson et al., 2004) and porcine insulin (Ishikawa et al., 2008).
Microseeding is normally exploited in obtaining X-ray diffraction quality crystals, although it might be useful for growing larger crystals, especially when it is necessary to reduce the number of nucleation sites per drop. In the microseeding procedure one would grow small crystals and then crush them to generated microscopic crystals that will be used later as seeds for growing larger crystals. In microseeding, it is important to make sure that the crystallization conditions achieved in the drop containing the seeds are located inside the metastable zone, where the crystals will grow from the seeds and new nucleation events will not happen. An excellent tool for making microseeds, called the Seed BeadTM, is supplied by Hampton Research Corp. The microseeding approach is not very popular in the world of the production of neutron diffraction quality crystals, even though it was reported as a means of growing large crystals of photoactive yellow protein (Yamagushi et al., 2009) and rubredoxin (Kurihara et al., 2004). The preferred seeding technique in neutron protein crystallography is macroseeding. Sometimes neither refining the crystallization conditions nor increasing the volume of the crystallization drops leads to large crystals, which stop growing reaching only submillimeter scales. In these cases, macroseeding may afford crystals that are a few fold bigger, leaping from unfeasible crystals to the ones worth collecting neutron diffraction data from. Macroseeding is often done repeatedly, working with one crystal - a macroseed - and increasing its size each time it is placed into a new crystallization drop. (N.B. Before the crystal is transferred to another crystallization drop it must be washed, at least twice, with the precipitant solution, which can be the mother liquor or well solution, and any visible small crystals attached to the macroseed have to be carefully removed). As was the case for microseeding, the conditions in the drop containing the macroseed should correspond to the metastable zone to avoid the nucleation of new microcrystals. This is achieved generally by lowering the [protein] in the drop to which the macroseed is introduced. Several neutron structures have been determined from the protein crystals grown by the macroseeding approach, including amicyanin (Sukumar et al., 2005), elastase (Kinoshita et al., 2007) and HIV-1 protease/inhibitor (Matsumura et al., 2008)
Temperature is known to influence the solubility and, thus, the crystallizability of proteins. With the few exceptions, like Na2SO4, from the small-molecule world, the solubility of a protein drops with lowering the temperature. Therefore, altering temperature during crystallization can be used to one's advantage for obtaining larger crystals. The protein stability at room temperature (RT) should be carefully examined; the crystal growth might have to be carried out at refrigerating temperatures of 4-6°C, without the possibility of changing it. If the protein under consideration is stable at RT it is advisable to start crystallization at ~20°C (or at lower temperature if the stability is a concern). When the enlargement in the crystal size is no longer noticeable, one should initiate a sequence of lowering the temperature by 1-2°C every week or so until it reaches ~6°C, thereby maintaining the conditions within the metastable (crystal growth) zone. Many proteins, once crystallized, are stable at 6°C and may continue to grow for months. Understandably, if one's protein is not particularly stable at RT, the whole crystallization procedure might have to be done at 4°C without any temperature alterations. The technique of slow cooling was successfully applied to grow large crystals of HIV-1 protease/inhibitor complex (Matsumura et al., 2008) and urate oxidase (Oksanen et al., 2009).
Crystal mounting for neutron diffraction. Neutron diffraction studies are generally carried out at room temperature, which is advantageous for protein function and structural enzymology studies. Therefore, protein crystals are always packed into quartz capillaries and sealed air-tight with wax. Unlike in X-ray crystallography, the quartz capillaries used for neutron diffraction can be thick-walled. In fact, capillaries with wall thickness up to 0.4 mm are acceptable because thermal neutrons are scattered weakly by quartz. It is also useful to shape the capillaries into the hourglass form with CH4/O2 torch and position a crystal at the narrow waist of the tube, as illustrated on Figure 2. This will ensure that the crystal does not have any freedom to move during the data collection. The capillary waist is to be left with ~1 mm inner diameter to avoid traveling of the mother liquor to the crystal when the capillary is tilted or even turned upside down.
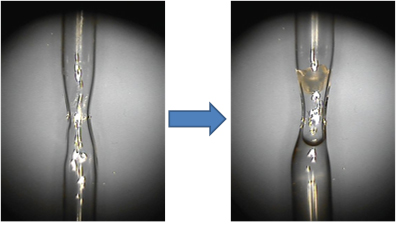
Figure 2. The empty quartz capillary shaped as hourglass (left) and the capillary containing a crystal above and the deuterated mother liquor below the narrow waist (right). The crystal is secured by the narrowing capillary, while H/D exchange occurs by vapor-diffusion due to the presence of the D2O solution underneath the crystal.
H/D exchange. To improve the signal-to-noise ratio in a neutron diffraction experiment it is advantageous to substitute hydrogen atoms with deutreium atoms in the protein molecule, i.e. perform H/D exchange. One way to accomplish this is to grow a crystal in solutions made exclusively with heavy water (D2O) so that hydrogen atoms at all exchangeable sites, i.e. in the functional groups OH, NH and SH, and solvent water molecules, are substituted with deuterium. This approach has been successfully exploited for growing neutron diffraction quality crystals of, e.g., amicyanin (Sukumar et al., 2010), HIV-1 protease/inhibitor (Matsumura et al., 2010) and elasatase/inhibitor complexes (Tamada et al., 2009), photoactive yellow protein (Yamaguchi et al., 2009), thaumtin (Teixeira et al., 2008), dissimilatory sulfite reductase D (Chatake et al., 2003) and of a few other proteins. Although, crystallization conditions with deuterated buffers and precipitating agents should be considered, these conditions may significantly differ from those with H2O solutions, resulting in much more effort needed to grow desired crystals. In addition, the protein crystals may not grow as large as they did when hydrogenous conditions were employed. This consideration holds true also for many per-deuterated proteins, in which all hydrogen atoms are replaced with deuterium by expressing the protein in per-deuterated media. Empirical experience demonstrates that crystallization of a protein for neutron diffraction should start in the hydrogenous conditions. If these crystallization efforts fail to produce large crystals, per-deuteration is advised, since per-deuterated crystals may sometimes grow as large as hydrogenous, while the effective neutron diffraction from the former will be a few fold stronger than from the latter. On the other hand, examination of the statistics of the deposited neutron structures to the Protein Data Bank shows that out of 41 neutron structures deposited to date (July 2010) only three proteins, rubredoxin (Gardberg et al., 2010), sperm whale myoglobin (Shu, Ramakrishnan & Schoenborn, 2000) and human aldose reductase (Blakeley et al., 2008), are per-deuterated. The success rate for obtaining a neutron structure from a per-deuterated crystal is, therefore, under 10%. As a consequence, one should undoubtedly investigate other options before embarking on a journey to the per-deuteration.
Protein crystals can also be first grown from fully hydrogenous solutions and the H/D exchange be completed later by vapor diffusion inside the quartz capillary (Figure 2). This technique is less invasive and complex than growing crystals in D2O, while resulting in similar H/D exchange levels provided sufficient time of ~ 1 month would have passed before neutron diffraction experiment commenced. The H/D exchange occurs by the D2O vapor that comes from the mother liquor plug and readily penetrates the solvent-rich protein crystal.
Cited Literature.
Blakeley, M. P., Ruiz, F., Cachau, R., Hazemann, I., Meilleur, F., Mitschler, A., Ginell, S., Afonine, P., Ventura, O. N., Cousido-Siah, A., Haertlein, M., Joachimiak, A., Myles, D. & Podjarny, A. 2008. Proc. Natl. Acad. Sci. USA 105, 1844-1848.
Budayova-Spano, M., Bonnete, F., Ferte, N., El Hajji, M., Meilleur, F., Blakeley, M. P. & Castro B. 2006. Acta Crystallogr. F62, 306-309.
Chatake, T., Mizuno, N., Voordouw, G., Higuchi, Y., Arai, S., Tanaka, I. & Niimura, N. 2003. Acta Crystallogr. D59, 2306-2309.
Chatake, T., Shibayama, N., Park, S.-Y., Kurihara, K., Tamada, T., Tanaka, I., Niimura, N., Kuroki, R. & Morimoto, Y. 2007. J. Am. Chem. Soc. 129, 14840-14841.
Fisher, S. Z., Kovalevsky, A. Y., Domsic, J. F., Mustyakimov, M., McKenna, R., Silverman, D. N. & Langan P. A. 2010. Biochemistry 49, 415-421.
Gardberg, A. S., del Castillo, A. R., Weiss, K. L., Meilleur, F., Blakeley, M. P. & Myles, D. A. A. 2010. Acta Crystallogr. D66, 558-567.
Hanson, B. L., Langan, P., Katz, A. M., Li, X., Harp, J. M., Glusker, J. P., Schoenborn, B. P. & Bunick, G. J. 2004. Acta Crystallogr. D60, 241-249.
Ishikawa, T., Chatake, T., Ohnishi, Y., Tanaka, I., Kurihara, K., Kuroki, R. & Niimura, N. 2008. Chem. Phys. 345, 152-158.
Kinoshita, T., Tamada, T., Imai, K., Kurihara, K., Ohhara, T., Tada, T. & Kuroki, R. 2007. Acta Crystallogr. F63, 315-317.
Kovalevsky, A. Y., Hanson, L., Fisher, S. Z., Mustyakimov, M., Mason, S. A., Forsyth, V. T., Blakeley, M. P., Keen, D. A., Wagner, T., Carrell, H. L., Katz, A. K., Glusker, J. P. & Langan, P. 2010a. Structure 18, 688-699.
Kovalevsky, A. Y., Seaver, S., Fisher, S. Z., Mustyakimov, M., Sukumar, N., Langan, P., Mueser T. C. & Hanson, B. L. 2010b. Acta Crystallogr. F66, 474-477.
Kurihara, K., Tanaka, I., Chatake, T., Adams, M. W. W., Jenney, Jr., F. E., Moiseeva, N., Bau, R. & Niimura, N. 2004. Proc. Natl. Acad. Soc. 101, 11215-11220.
Matsumura, H., Adachi, M., Sugiyama, S., Okada, S., Yamakami, M., Tamada, T., Hidaka, K., Hayashi, Y., Kimura, T., Kiso, Y., Kitatani, T., Maki, S., Yoshikawa, H. Y., Adachi, H., Takano, K., Murakami, S., Inoue, T., Kuroki, R. & Mori, Y. 2008. Acta Crystallogr. F64, 1003-1006.
Oksanen, E., Blakeley, M. P., Bonnete, F., Dauvergne, M. T., Dauvergne, F. & Budayova-Spano, M. 2009. J. R. Soc. Interface 6, S599-S610.
Shu, F., Ramakrishnan, V. & Schoenborn B. P. 2000. Proc. Natl. Acad. Sci. USA 97, 3872-3877.
Sukumar, N., Langan, P., Mathews, F. S., Jones, L. H., Thiyagarajan, P., Schoenborn, B. P. & Davidson, V. L. 2005. Acta Crystallogr. D61, 640-642.
Sukumar, N., Mathews, F. S., Langan, P. & Davidson, V. L. 2010. Proc. Natl. Acad. Sci. USA 107, 6817-6822.
Tamada, T., Kinoshita, T., Kurihara, K., Adachi, M., Ohhara, T., Imai, K., Kuroki, R. & Tada, T. 2009. J. Am. Chem. Soc. 131, 11033-11040.
Teixeira, S. C. M., Blakeley, M. P., Leal, R. M. F., Mitchell, E. P. & Forsyth, V. T. 2008. Acta Crystallogr. F64, 378-381.
Yamaguchi, S., Kamikubo, H., Kurihara, K., Kuroki, R., Niimura, N., Shimizu, N., Yamazaki, Y. & Kataoka, M. 2009. Proc. Natl. Acad. Sci. USA 106, 440-444.
